
причины, симптомы, диагностика и лечение

Липоидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов и их метаболитов в различных органах и тканях. Общие клинические проявления представлены прогрессирующим расстройством интеллектуальных и двигательных функций, поражением костей, кожи, центральной нервной системы, глаз и внутренних органов (печени, почек, селезенки). Диагностика основана на лабораторных исследованиях ферментативной активности, количества токсичного субстрата, наличия мутации в генах. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
Общие сведения
Синонимичные названия липоидозов – липидозы, ретикулоэндотелиозы, лизосомные болезни накопления липидов. К данной группе относится сфингомиелиноз, болезнь Тея-Сакса, болезнь Гоше, семейные гиперлипидемии и некоторые другие заболевания. Общей характеристикой является патологическое накопление липидов внутри клеток организма в результате дефекта ферментных систем. Липоидозы относятся к группе редких (орфанных) заболеваний. Их распространенность очень низка, для отдельных типов патологий составляет от 1:40 тыс. до 1:1 млн. и реже. Суммарная частота – 1:7 тыс. Большинство липидозов имеют прогредиентное течение, приводят к инвалидизации и ранней смерти.

Липоидозы
Причины липоидозов
Определяющим фактором развития липидозов является генетический дефект, который обуславливает полную или частичную недостаточность лизосомальных ферментов, расщепляющих сложные липиды. Наследование болезней происходит по аутосомно-рецессивному механизму. Это означает, что новорожденный оказывается больным, если получает мутационный ген от каждого родителя (имеет пару дефектных генов в аллели). Когда мутация передается только от матери или отца, то ребенок является ее носителем и остается здоровым.
Исключительный механизм передачи дефекта у болезни Фабри. В отличие от других липидозов, она наследуется по X-сцепленному рецессивному типу. Гемизиготные пациенты мужского пола больны, передают мутацию только дочерям. У девочек заболевание всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза проявляются у пациенток с одним дефектным геном, когда доминантный ген оказывается инактивированным (причины этого не выяснены).
Патогенез
Патогенетической основой большинства липоидозов является генетически детерминированный ферментативный дефект – энзимопатия. Вследствие этого лизосомы клеток накапливают липиды и промежуточные продукты их метаболизма, что ведет к прогрессивному нарастанию нарушения функций органов. При синдроме Вольмана определяется недостаточность кислой этеразы, становится невозможным полный цикл обмена холестерола, повышается содержание его эфиров в лизосомах селезенки, печени, надпочечников, кишечника и костного мозга. У пациентов с болезнью Гоше снижено или полностью отсутствует производство бета-глюкозидазы; в печени, селезенке, костном мозге скапливаются продукты расщепления сфинголипидов. Болезнь Нимана-Пика развивается на базе дефицита сфингомиелазы, характеризуется повышением количества сфингомиелина во многих клетках, особенно в гепатоцитах, нейронах. При идиотии Тэя-Сакса существует дефект N-ацетилгексозаминидазы, происходит накапливание ганглиозидов в головном мозге.
Классификация
Наследственные нарушения метаболизма сложных липидов представлены группой различных по происхождению заболеваний, которые на патогенетическом уровне связаны с патологией жирового обмена. Для липоидозов характерно скопление сложных липидных соединений внутри лизосом клеток. В зависимости от того, какой липид не расщепляется до конца и откладывается в тканях, выделяют несколько типов болезней:
- Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов – соединений, состоящих из углеводов и жирных кислот. Гликолипидозы представлены цереброзидозами (сфинголипидоз Гоше, болезнь Краббе), сульфатидозами (метахромотическая лейкодистрофия), церамидолигозидозами (болезнь Фабри, церамидлактозидлипоидоз), ганглиозидозами (болезнь Сандхоффа, ранняя детская амавротическая идиотия).
- Липопротеинемии. Обусловлены патологией обмена липидов плазмы крови, основанной на генетическом дефекте ферментов или рецепторов клеток. Липиды плазмы – жирные кислоты, триглицериды, холестерин. Липопротеинемии включают семейную гиперхолестеринемию, комбинированную семейную гиперлипидемию и семейные гиперлипидемии типов I-V.
- Сфингомиелиноз. Синонимичное название – болезнь Ниманна-Пика. При развитии этого заболевания в ретикулоэндотелиальных клетках увеличивается содержание фосфолипидного соединения сфингомиелина.
Симптомы липоидозов
Клиническая картина липидозов определяется особенностями вовлечения в патологический процесс органов и систем. При болезни Гоше I типа поражается печень, селезенка, кости и костный мозг. Симптомы включают увеличение размеров печени, хрупкость костей, анемию, лейкопению, снижение свертываемости крови. II тип заболевания разворачивается с преимущественным повреждением ЦНС и печени. Наблюдаются судорожные приступы, мышечный гипертонус, спастичность, интеллектуальная недостаточность, расстройства акта глотания. У людей с галактозилцерамидным липидозом (болезнью Краббе) снижается функциональность миелиновой оболочки. Развивается гипервозбудимость, рвота, судороги, задержка психомоторного развития с прогрессирующим снижением интеллекта и зрения.
При метахромотической лейкодистрофии патологически изменяется миелиновая оболочка. Основные симптомы – гипотония мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, глухота, интеллектуальное и моторное недоразвитие. Клинические признаки болезни Андерсона-Фабри разнообразны, одними из наиболее распространенных являются нейропатическая боль (умеренной и легкой выраженности, в ступнях и ладонях) и кризы Фабри – сильные жгучие боли в конечностях приступообразного характера. Дополнительно возникает гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератомы, расстройства слуха, сердечно-сосудистой и почечной функций.
При развитии синдрома Сандхоффа определяется общая вялость, гипотонус мышц конечностей, трудности сосания и глотания, прогрессирующая задержка моторного и психического развития, двигательная слабость, шумы в сердце, судороги, слепота, увеличение селезенки. При ранней детской амавротической идиотии больше всего поражается центральная нервная система. К концу первого полугодия ухудшается реакция детей на внешние сигналы (лица близких, игрушки), утрачиваются двигательные навыки, снижается познавательный и игровой интерес. Нарушается зрение вплоть до слепоты. Формируются судорожные припадки.
Первые симптомы сфингомиелиноза – вялость, малоподвижность, апатичность, отказ от еды, рвота. Позже увеличивается живот (гепатомегалия), конечности становятся худыми, кожа приобретает коричневатый оттенок, периоды заторможенности сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Диагностируется умеренная гидроцефалия, гипертермия, спастический парез ног и рук, приступообразная асфиксия.
Проявления гиперлипопротеинемий обнаруживаются в возрасте до 10 лет. Для всех форм болезней свойственно отложение подкожного жира в виде ксантом, а также абдоминальные боли, панкреатит, гепатоспленомегалия (скопление липидов в селезенке, печени). При комбинированной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз сосудов. При гиперлипидемиях IV и V типа – снижение чувствительности к глюкозе, ИБС.
Осложнения
Липоидозы протекают с развитием недостаточности жизненно важных органов. Наиболее частыми осложнениями становятся заболевания сердца и сосудов, ЦНС, почек, легких, печени. Пациенты страдают от атеросклероза сосудов, сердечной и дыхательной недостаточности, ишемической болезни сердца, хронической почечной и печеночной недостаточности, аденом и цирроза печени, инсультов, транзиторных ишемических атак. При типах заболеваний, совместимых с жизнью, зачастую нарушено двигательное и психическое развитие. Многие больные оказываются неспособны к самообслуживанию, обучению и овладению профессией, нуждаются в пожизненном уходе со стороны окружающих.
Диагностика
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением занимаются педиатры, гематологи, гастроэнтерологи, ревматологи, неврологи, психиатры и врачи-генетики. На первичном этапе диагностики проводится сбор семейного анамнеза: при наследственном характере ферментопатии у больного могут быть родственники с подтвержденным диагнозом липидоза. При сборе клинических данных специалисты обращают внимание на время начала симптомов: чаще всего болезнь проявляется в период новорожденности или первого года жизни, редко – у детей постарше или взрослых. К специфическим методам обследования пациентов относят:
- Анализ активности дефектного фермента. Исследованию подвергаются различные биоматериалы – плазма крови, лейкоциты, сухие пятна крови, культура фибробластов кожи, биопсийный материал почек, печени. При липоидозах определяется недостаток активности определенного фермента: от легкого снижения до полного отсутствия.
- Количественное исследование липидов. В крови и биопсийном материале органов анализируется содержание патологически накапливаемых липидов и промежуточных продуктов их обмена. У больных липоидозом показатели превышают норму. Параллельно изучаются изменения строения пораженных клеток.
- Секвенирование ДНК. Выявление дефектного гена в хромосоме является наиболее точным, но трудоемким методом диагностики наследственных болезней. Широко применяется в рамках перинатальной и преимплантационной диагностики, в случаях, когда вышеперечисленные анализы не дают однозначного представления о диагнозе.
- Визуализирующие исследования органов. Дополнительно проводится диагностика состояния пораженных органов – сердца, печени, желчного пузыря, селезенки, легких, почек, головного мозга. Используется УЗИ, МРТ, КТ, ЭКГ, ЭЭГ. Процедуры позволяют оценить размеры, выявить структурные изменения и новообразования в органе.
Лечение липоидозов
Терапия данной группы заболеваний – сложная задача для врачей разных специальностей. Методы лечения несовершенны и продолжают разрабатываться, при некоторых видах болезней возможно добиться лишь временного улучшения самочувствия больного, при других достижима стойкая ремиссия. Общая схема медицинской помощи больным состоит из трех компонентов:
- Ферментозаместительная терапия. В организм пациентов вводятся препараты с искусственно выделенным дефицитарным ферментом. Инъекции выполняются пожизненно, позволяют восстановить метаболизм липидов.
- Субстратредуцирующая терапия. Лечение направлено на снижение интенсивности образования патологически накапливаемого соединения (сложного липида, его метаболитов). Используются молекулы с низкой массой, которые стимулируют остаточную активность фермента.
- Симптоматическая терапия. Препараты подбираются индивидуально на основании клинической картины болезни. Распространено применение антиконвульсантов, обезболивающих средств, ингибиторов АПФ, гепатопротекторов. При отдельных видах липоидозов использование симптоматических лекарств является единственным способом лечения.
Прогноз и профилактика
Липоидозы характеризуются гетерогенностью генетического дефекта и выраженной клинической полиморфностью. Большинство из них имеет непрерывное прогрессирующее течение. Правильная диагностика и своевременное лечение позволяют увеличить продолжительность жизни больных, а при легких формах способствуют улучшению адаптации. Профилактика возможна на этапе планирования и в первые месяцы беременности. Супружеским парам с высоким риском передачи болезни ребенку рекомендуется медико-генетическое консультирование, а в первом триместре – исследование амниотической жидкости и материала биопсии хориона на наличие мутаций генов.
www.krasotaimedicina.ru
Липидоз — это… Что такое Липидоз?
липидоз — сущ., кол во синонимов: 1 • липоидоз (1) Словарь синонимов ASIS. В.Н. Тришин. 2013 … Словарь синонимов
липидоз — (lipidosis; липиды + оз) 1) (липоидоз устар.) общее название наследственных или приобретенных болезней, характеризующихся нарушением липидного обмена; 2) см. Дистрофия жировая … Большой медицинский словарь
Липидоз (Lipidosis), Липоидоз (Lipoidosis)
ЛИПИДОЗ (LIPIDOSIS), ЛИПОИДОЗ — (lipoidosis) нарушение внутриклеточного липидного обмена в организме. Церебральный липидоз (brain lipidoses) (см. Болезнь Гоше, Синдром Гунтера, Синдром Гурлера, Болезнь Тэя Сакса) является врожденным нарушением внутриклеточного липидного… … Толковый словарь по медицине
фосфатидный липидоз
Липидо́зы — (lipidoses; липид [ы] (Липиды) + ōsis) группа заболеваний, характеризующихся нарушением липидного обмена и имеющих преимущественно наследственный характер. Большинство Л. относится к болезням накопления (Болезни накопления), которые обусловлены… … Медицинская энциклопедия
Тилорон — (Tiloronum) Химическое соединение … Википедия
болезнь Ниманна-Пика
дистрофия жировая — (d. adiposa; син.: декомпозиция жировая, Д. липидная, липидоз, липофанероз, ожирение дегенеративное, ожирение декомпозиционное, фанероз жировой, фетофанероз) Д., характеризующаяся нарушением обмена липидов и проявляющаяся либо увеличением… … Большой медицинский словарь
липоидоз — (устар.; lipoidosis; липоиды + оз) см. Липидоз … Большой медицинский словарь
Дистрофия — I Дистрофия у детей (dystrophia; греч. dys + trophē питание) хронические расстройства питания у детей. Различают следующие основные виды дистрофии: гипотрофию, гипостатуру, паратрофию и гипертрофию (см. Ожирение). Кроме того, выделяют особый… … Медицинская энциклопедия
dic.academic.ru
ЛИПИДОЗЫ — Большая Медицинская Энциклопедия
ЛИПИДОЗЫ (lipidosis; липид(ы) + -osis; устар. липоидозы) — группа заболеваний, характеризующихся нарушением липидного обмена и носящих в большинстве случаев наследственный характер. Причиной наследственных (первичных) Л. чаще бывает генетический дефект какого-либо фермента. Большинство Л. является болезнями накопления (см. Тезаурисмозы), т. к. в результате недостаточности определенного фермента, участвующего в обмене липидов (см. Жировой обмен) или липопротеидов (см.), в клетках или биол, жидкостях организма обнаруживают аномально большие количества нерасщепленного субстрата соответствующего фермента. Классификация окончательно не разработана, механизмы возникновения Л. продолжают изучаться.
Большая группа Л.— гликолипидозы (см. Гликозидозы), т. е. заболевания, в основе патогенеза которых лежит нарушение распада углеводного компонента гликолипидов (см.). Наиболее распространенные гликолипиды в организме человека — сфингогликолипиды, поэтому заболевания, связанные с нарушением обмена углеводной части их молекул, носят самостоятельное название «сфингогликолипидозы». Таким образом, термины «гликолипидозы» и «сфингогликолипидозы» близки. Однако, строго говоря, эти термины не являются синонимами широко используемого в литературе термина «сфинголипидозы», объединяющего более широкую группу наследственных болезней с нарушением обмена (в основном распада) не только углеводного, но и липидного компонента липидов этого класса. Несмотря на то, что большинство известных сфинголипидозов представляет собой сфингогликолипидозы, к этой же группе болезней относятся заболевания, при которых генетически обусловлено отсутствие ферментов, катализирующих распад липидного компонента сфингогликолипидов (напр., при болезни Фарбера отсутствует фермент церамидаза) или катализирующих отщепление остатков неуглеводной природы, таких как фосфохолин (напр., сфингомиелиназа при болезни Ниманна—Пика) или сульфат (напр., арилсульфатаза А при метахроматической лейкодистрофии).
Наследственно обусловленное нарушение обмена ганглиозидов (гликолипидов, углеводная часть молекулы которых содержит остаток сиаловой кислоты) является причиной ганглиозидозов. К ганглиозидозам относится болезнь Тея—Сакса (GM2-ганглиозидоз, тип I), в мозге больных этой болезнью накапливаются значительные количества Ом2-ганглиозида в отсутствие гексозаминидазы А, для к-рой Gm2-ганглиозид является субстратом (см. Амавротическая идиотия). При так наз. нулевом варианте болезни Тея—Сакса — болезни Сандгоффа (Gm2-ганглиозидозе II типа) в тканях больных отсутствуют активности гексозаминидаз А и В. Кроме болезни Сандгоффа, различают юношеский GM2-ганглиозидоз III типа, или болезнь Бернгеймера—Зайтельбергера, характеризующийся уменьшением активности гексозаминидазы А, и инфантильный (детский) GM2-ганглиозидоз, вариант АВ, при к-ром активность гексозаминидаз А и В не изменяется, но отсутствует специфический термолабильный фактор, необходимый для расщепления Gm2-ганглиозида гексозаминидазой А. Этот тип GM2-ганглиозидоза может быть выявлен только в том случае, если в качестве субстрата используется природный GM2-ганглиозид, т.к. на расщепление синтетического субстрата специфический белковый фактор влияния не оказывает. К частичным ганглиозидозам, связанным с нарушением распада GM1-ганглио-зида, относятся генерализованный GM1-ганглиозидоз, или болезнь Нормана—Ландинга (отсутствуют бета-галактозидазы А, В и С), и юношеский GM1-ганглиозидоз, или болезнь Дерри (значительно снижена активность В- и С-форм бета-галактозидазы).
Имеются сведения о новом типе гликолипидоза — GM3-ганглиозидозе, развитие к-рого связано не с накоплением нерасщепившихся ганглиозидов, т. е. с генетически обусловленным блоком их катаболизма, а с нарушением их биосинтеза. Накопление в печени и мозге больных этой болезнью GM3-ганглиозида происходит в результате отсутствия специфической N-ацетилгалактозаминилтрансферазы, катализирующей превращение GM3-ганглиозида в GM2-ганглиозид.
Болезнь Фабри — единственная среди гликолипидозов, при к-рой генетический дефект определенно связан с X-хромосомой; она обусловлена дефицитом активности церамидтригексозид-альфа-галактозидазы А (см. Фабри болезнь). При болезни Фабри в тканях больных, а также в их лейкоцитах и фибробластах накапливается большое количество церамидтригексозида, который выводится с мочой.
Один из очень распространенных гликолипидозов — болезнь Гоше (см. Гоше болезнь). Основным продуктом накопления при ней является церамидглюкозид (глюкоцеребреброзид) — не расщепленный субстрат реакции, катализируемой кислой бета-глюкозидазой (глюкоцереброзидазой), который накапливается в ретикулоэндотелиальных клетках. Активность кислой бета-глюкозидазы (см. Глюкозидазы) при этой болезни снижается в разной степени в зависимости от формы болезни вплоть до полного отсутствия. Для наиболее тяжелой инфантильной формы болезни остаточная активность глюкоцереброзидазы составляет менее 9% от активности фермента в норме. Юношеская форма болезни характеризуется 10 — 17% остаточной активности глюкоцереброзидазы, а взрослая форма — даже 40% активности фермента. Существование различных форм болезни Гоше обусловлено, по-видимому, различными мутациями в одном и том же (или сходных) генетическом локусе, контролирующем образование глюкоцереброзидазы.
В основе патогенеза глобоидноклеточной лейкодистрофии (болезни Краббе) и семейной метахроматической лейкодистрофии Гринфилда— Шольца (см. Лейкодистрофии) лежит генетически детерминированный дефицит бета-галактозидазы (церамидгалактозидазы) и цереброзидсульфатазы (арилсульфатазы А). При болезни Краббе продуктом накопления является церамидгалактозид (галактоцереброзид), а при семейной метахроматической лейкодистрофии в нервных клетках, нервных волокнах, в глие ц. н. с., а также в сетчатке глаз, канальцах ночек и лейкоцитах накапливаются цереброзид-сульфатиды.
По клинической картине на болезнь Краббе похож муколипидоз, однако при нем объем внутренних органов не увеличивается и количество мукополисахаридов в моче остается в норме.
При генетически обусловленном дефекте сфингомиелиназы в ретикуломакрофагальной системе и головном мозге, а также в мезенхимальных и эпителиальных элементах многих органов накапливается большое количество сфингомиелина. В костном мозге появляются многоядерные макрофаги — клетки Пика (см. Ниманна-Пика болезнь).
При гаргоилизме (см.) и других мукополисахаридозах (см.) также отмечают значительные изменения в обмене липидов, преимущественно гликолипидов, холестерина и фосфолипидов .
В 1888 г. американский невропатолог и психиатр Деркум (F. X. Dercum) впервые описал нейроэндокринное заболевание, характерным проявлением к-рого было образование под кожей множественных болезненных липом. Предполагают, что эта болезнь носит наследственный характер, т. к. ее наблюдали у нескольких членов одной семьи (см. Деркума болезнь ).
При генетическом дефекте панкреатической липазы утрачивается способность полностью переваривать жиры. Ок. 50—80% жиров, поступающих с пищей, выделяется с калом, который отличается маслянистой консистенцией. Недостаточность кислой липазы является причиной болезни Вольмана, при к-рой во внутренних органах обнаруживаются «пенистые» клетки, содержащие большое количество триглицеридов и холестерина. Заболевание характеризуется обызвествлением надпочечников, гепатоспленомегалией и ксантоматозом (см.), т. е. отложением в коже и некоторых тканях холестерина и триглицеридов, проявляющемся образованием ксантом (см.).
Ксантоматоз сопровождает и липопротеинемию I типа [по Фредриксону (D. S. Fredrickson)], развивающуюся вследствие генетического дефекта липопротеидлипазы (см.). Наследственная аальфалипопротеинемия — танжерская болезнь — характеризуется отсутствием в крови нормальных липопротеидов высокой плотности (α-липопротеидов) и появлением аномальных липопротеидов высокой плотности (см. Липопротеиды).
Другая наследственная патология такого типа — абеталипопротеинемия (см.) характеризуется отсутствием в крови не только бета-липопротеидов, но и пре-бета-липоиротеидов и хиломикронов. В этом случае главный метаболический дефект связан с угнетением синтеза белка апо-В в печени. Этот белок необходим для формирования перечисленных липопротеидов. Известно наследственное заболевание, вызванное недостаточностью лецитин: холестерин — ацилтрансферазы (так наз. ЛХАТ-недостаточность). Этот фермент катализирует этерификацию холестерина. В результате нарушения этерификации холестерина состав и морфология липопротеидов плазмы крови изменяются. Для этого заболевания характерно отложение неэтерифицированного холестерина в селезенке, почках, роговице, эритроцитарной мембране. Клинические проявления его — гипохромная анемия, почечная недостаточность, спленомегалия, помутнение роговицы и т. д. Кроме того, в крови появляются аномальные липопротеиды-X.
Приобретенные (вторичные) Л. обычно связаны с нарушением обмена холестерина. К ним относятся заболевания опухолевой природы, получившие название гистиоцитозов (см.), а также вторичный гиперхолестеринемический ксантоматоз и вторичный гиперлипемический ксантоматоз. В группу гистиоцитозов входят острый системный прогрессирующий гистиоцитоз (болезнь Леттерера—Сиве), хрон, системный прогрессирующий гистиоцитоз (болезнь Хенда—Шюллера—Крисчена) и эозинофильная гранулема. Болезнь Xенда—Шюллера—Крисчена напоминает некоторые сфинголипидозы. В основе патол, процесса при этой болезни лежит нарушение обмена холестерина. Клин, проявление ее — ксантоматоз с дистрофией костной ткани и внутренних органов, вызванной отложением холестерина и его эфиров, триглицеридов и фосфатидов в пролиферирующих гистиоцитах, приобретающих пенистый вид и превращающихся в многоядерные гигантские макрофаги.
Местным проявлением ряда патол, процессов и болезней являются вторичные локальные Л., которые могут быть связаны с деструкцией тканей, богатых липидами, с образованием аномальных количеств липидов при тех или иных патол, процессах, с местным расстройством крово- и лимфообращения, с воспалением или развитием опухоли. Морфол, признаками таких Л. являются образование липофагов и липогранулем, липосклеротических (атеросклеротических) бляшек п атером, липоидная инфильтрация как вновь образующихся субстанций (гиалин, амилоид), так и тканей при воспалении, репаративной регенерации или опухоли. Наибольшее распространение среди вторичных локальных Л. имеет атеросклероз (см.).
Экспериментальный сосудистый липидоз
По отдельным морфол, и цитол, признакам к Л. может быть отнесен так наз. экспериментальный сосудистый Л., при к-ром нарушается обмен холестерина и липопротеидов; механизм этих нарушений не выяснен. Экспериментальный сосудистый Л. по нек-рым данным представляет собой начальную стадию атеросклероза.
Экспериментальный сосудистый Л. может быть получен в результате скармливания животным больших количеств холестерина, насыщенного жира, а также после многократного внутривенного введения животным гомологичных липопротеидов низкой и очень низкой плотности, предложенного А. Н. Климовым и соавт. (1967). Чаще всего для этой цели используют кроликов, свиней миниатюрной породы и обезьян; в процессе эксперимента у них отмечается нарастание в плазме крови концентрации липопротеидов низкой и очень низкой плотности.
Морфологически экспериментальный сосудистый липидоз прежде всего проявляется в образовании небольших липидных пятен и полосок желтоватого цвета. В пятнах и полосках резко увеличено содержание холестерина, в меньшей степени фосфолипидов и триглицеридов.
Последовательность изменений, происходящих в сосудистой стенке, представляется следующим образом. Вначале проявляются изменения проницаемости эндотелиального барьера. В ответ на увеличение уровня бета- и пре-бета-липоиротеидов в крови повышается пиноцитозная активность эндотелиальных клеток. Одновременно с этим происходят изменения на поверхности эндотелиальных клеток — разрыхление и исчезновение защитного углеводного слоя (гликокаликса). Повышение пиноцитозной активности сопровождается частичным или полным раскрытием межэндотелиальных пространств, что позволяет беспрепятственно проникать и откладываться во внутренней оболочке артерий липопротеидам и другим белкам плазмы крови. Этому способствует образование в сосудистой стенке физ.-хим. комплекса бета- и пре-бета-липопротеидов и гликозаминогликанов основного вещества ее внутренней оболочки. Заполнение липидными вакуолями цитоплазмы эндотелиальных клеток, их сокращение или растяжение набухающей межуточной тканью вызывает гибель отдельных эндотелиальных клеток.
На этой стадии макроскопически липидные пятна еще не видны, а микроскопически наблюдается диффузное отложение липидов только в основном веществе.
В ответ на поступление в субэндотелиальный слой липопротеидов отмечается реакция со стороны основного вещества и клеток, его продуцирующих : увеличивается количество кислых гликозаминогликанов, усиливается образование коллагеновых и эластических волокон. Клеточная реакция выражается прежде всего в усиленной пролиферации гладкомышечных клеток. Гладкомышечные клетки субинтимального слоя средней оболочки артерии (миоинтимальные пенистые клетки) проникают во внутреннюю оболочку через разрыхленную внутреннюю эластическую мембрану и принимают участие в поглощении липопротеидов и липидов.
Важным моментом метаболизма липидов в цитоплазме пенистых клеток является контакт и сплавление липидных вакуолей с первичными лизосомами (см.), благодаря чему образуются вторичные лизосомы. Часть продуктов гидролиза липопротеидов (жирные к-ты, аминокислоты и др.) путем диффузии через мембрану вторичной лизосомы поступают в цитоплазму клетки, где используются либо для энергетических нужд, либо для синтеза новых макромолекул. Свободный неэстерифицированный холестерин накапливается в цитоплазме клеток, часто в виде кристаллов, окруженных уплотненной цитоплазмой. Эфиры холестерина из-за их большей гидрофобности формируют подобно триглицеридам внутриклеточные капли. Резко сниженная активность холестеринэстеразных энзимов в цитоплазме клеток приводит к избыточному накоплению холестерина в клетках и артериальной стенке в целом.
При прогрессировании Л. может иметь место гибель перегруженных липидами клеток. Выход гидролитических ферментов в межклеточное пространство вызывает некроз окружающих тканей и способствует усиленной пролиферации соединительной ткани. Этому же сопутствует понижение активности окислительно-восстановительных ферментов в сосудистой стенке. Т. о. представляется возможным переход Л. в стадию образования атеросклеротических бляшек.
При кратковременном кормлении животных холестерином на стадии экспериментального сосудистого Л. могут иметь место процессы регрессии липидных очагов.
Библиография: Давиденкова Е. Ф. и Либерман И. С. Клиническая генетика, с. 285, Л., 1975; Маккьюсик В. А. Наследственные признаки человека, пер. с англ., М., 1976: Харрис Г. Основы биохимической генетики человека, пер. с англ., М., 1978, библиогр.; S р г a n g e r J. W. a. W i e e clin a n n H. R. The genetic mucolipidoses, diagnosis and differential diagnosis, Hum. Genet., v. 9. p. 113, 1970; Stanbu-r у J. B., W y n g a a r d e n J. B. a. Fredrick s o n D. S. The metabolic basis of inherited disease, N. Y. a. o., 1972.
B. B. Серов, H. Г. Будковская; А. H. Климов, B. А. Нагорнев (экспериментальный липидоз).
xn--90aw5c.xn--c1avg
липидоз — это… Что такое липидоз?
липидоз — сущ., кол во синонимов: 1 • липоидоз (1) Словарь синонимов ASIS. В.Н. Тришин. 2013 … Словарь синонимов
Липидоз (Lipidosis), Липоидоз (Lipoidosis) — нарушение внутриклеточного липидного обмена в организме. Церебральный липидоз (brain lipidoses) (см. Болезнь Гоше, Синдром Гунтера, Синдром Гурлера, Болезнь Тэя Сакса) является врожденным нарушением внутриклеточного липидного метаболизма,… … Медицинские термины
ЛИПИДОЗ (LIPIDOSIS), ЛИПОИДОЗ — (lipoidosis) нарушение внутриклеточного липидного обмена в организме. Церебральный липидоз (brain lipidoses) (см. Болезнь Гоше, Синдром Гунтера, Синдром Гурлера, Болезнь Тэя Сакса) является врожденным нарушением внутриклеточного липидного… … Толковый словарь по медицине
фосфатидный липидоз — Nieman Pick disease болезнь Ниманна Пика, фосфатидный липидоз. HЗЧ из группы липидозов, характеризуется увеличением печени, селезенки, лимфатических узлов, прогрессирующим исхуданием, наличием в кроветворных органах фосфатид позитивных крупных… … Молекулярная биология и генетика. Толковый словарь.
Липидо́зы — (lipidoses; липид [ы] (Липиды) + ōsis) группа заболеваний, характеризующихся нарушением липидного обмена и имеющих преимущественно наследственный характер. Большинство Л. относится к болезням накопления (Болезни накопления), которые обусловлены… … Медицинская энциклопедия
Тилорон — (Tiloronum) Химическое соединение … Википедия
болезнь Ниманна-Пика — фосфатидный липидоз HЗЧ из группы липидозов, характеризуется увеличением печени, селезенки, лимфатических узлов, прогрессирующим исхуданием, наличием в кроветворных органах фосфатид позитивных крупных вакуолизированных клеток, умственной… … Справочник технического переводчика
дистрофия жировая — (d. adiposa; син.: декомпозиция жировая, Д. липидная, липидоз, липофанероз, ожирение дегенеративное, ожирение декомпозиционное, фанероз жировой, фетофанероз) Д., характеризующаяся нарушением обмена липидов и проявляющаяся либо увеличением… … Большой медицинский словарь
липоидоз — (устар.; lipoidosis; липоиды + оз) см. Липидоз … Большой медицинский словарь
Дистрофия — I Дистрофия у детей (dystrophia; греч. dys + trophē питание) хронические расстройства питания у детей. Различают следующие основные виды дистрофии: гипотрофию, гипостатуру, паратрофию и гипертрофию (см. Ожирение). Кроме того, выделяют особый… … Медицинская энциклопедия
dic.academic.ru
— липидозы — Биохимия
Генетические заболевания, при которых происходит неполное расщепление полимерных веществ и их накопление, получили название лизосомные болезни накопления, так как они обусловлены дефектами специфических лизосомальных гидролаз. В случае накопления липидов такие болезни называются липидозы. При липидозах нарушается нормальный катаболизм липидов до соответствующих мономеров.
Липидозы
Болезнь Вольмана – редкое аутосомно-рецессивное заболевание из-за дефекта кислой эстеразы лизосом, что обусловливает накопление эфиров холестерола в лизосомах печени, селезенки, надпочечников, костного мозга и тонкого кишечника. Проявляется в первые недели жизни рвотой, диареей и стеатореей, гепатоспленомегалией и двусторонним кальцинозом надпочечников. Больные умирают в возрасте до 6 мес.
Болезнь Шюллера-Кристиана, аутосомно-рецессивное заболевание, характеризуется отложением в плоских костях, твердой мозговой оболочке и коже холестерола и его эфиров. Характерными являются деструктивные изменения в костях, остеопороз, мозжечковые расстройства. Заболевание обычно начинается в возрасте до 10 лет, реже позднее. Мужчины болеют в 2 раза чаще, чем женщины. Течение заболевания прогрессирующее. Дефектный фермент неизвестен.
Болезнь Гоше – отложение цереброзидов в макрофагальных клетках селезенки, печени, лимфатических узлов и костного мозга. Возникает в связи с аутосомно-рецессивным отсутствием гликоцереброзидазы (β-глюкозидазы). Основными симптомами заболевания являются спленомегалия, увеличение печени и селезенки, а также изменения в костях, проявляющиеся в виде остеопороза.
Дефектный фермент при болезни Гоше
При болезни Нимана-Пика наблюдается отложение сфингомиелина в клетках различных органов из-за дефицита сфингомиелиназы. Болезнь наследуется аутосомно-рецессивно, проявляется резким увеличением печени и селезенки, замедлением психического развития ребенка, появлением слепоты и глухоты. Чаще всего дети погибают в возрасте до 2 лет.
Дефектный фермент при болезни Нимана-Пика
Болезнь Тея-Сакса (амавротическая семейная идиотия) является результатом дефекта N-ацетилгексозаминидазы, при котором происходит отложение ганглиозидов в клетках головного мозга, что сопровождается атрофией зрительных нервов, слепотой, слабоумием и смертью в младенческом возрасте.
Дефектный фермент при болезни Тея-Сакса
biokhimija.ru
Липоидозы — Медицинский справочник
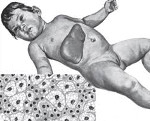
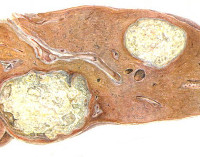
Липоидозы – наследственные заболевания, связанные с нарушением метаболизма жиров, отложением липидов и их метаболитов в различных органах и тканях. Общие клинические проявления представлены прогрессирующим расстройством интеллектуальных и двигательных функций, поражением костей, кожи, центральной нервной системы, глаз и внутренних органов (печени, почек, селезенки). Диагностика основана на лабораторных исследованиях ферментативной активности, количества токсичного субстрата, наличия мутации в генах. Лечение включает ферментозаместительную, субстратредуцирующую и симптоматическую терапию.
Липоидозы
Синонимичные названия липоидозов – липидозы, ретикулоэндотелиозы, лизосомные болезни накопления липидов. К данной группе относится сфингомиелиноз, болезнь Тея-Сакса, болезнь Гоше, семейные гиперлипидемии и некоторые другие заболевания. Общей характеристикой является патологическое накопление липидов внутри клеток организма в результате дефекта ферментных систем. Липоидозы относятся к группе редких (орфанных) заболеваний. Их распространенность очень низка, для отдельных типов патологий составляет от 1:40 тыс. до 1:1 млн. и реже. Суммарная частота – 1:7 тыс. Большинство липидозов имеют прогредиентное течение, приводят к инвалидизации и ранней смерти.
Причины липоидозов
Определяющим фактором развития липидозов является генетический дефект, который обуславливает полную или частичную недостаточность лизосомальных ферментов, расщепляющих сложные липиды. Наследование болезней происходит по аутосомно-рецессивному механизму. Это означает, что новорожденный оказывается больным, если получает мутационный ген от каждого родителя (имеет пару дефектных генов в аллели). Когда мутация передается только от матери или отца, то ребенок является ее носителем и остается здоровым.
Исключительный механизм передачи дефекта у болезни Фабри. В отличие от других липидозов, она наследуется по X-сцепленному рецессивному типу. Гемизиготные пациенты мужского пола больны, передают мутацию только дочерям. У девочек заболевание всегда проявляется при наличии двух рецессивных (измененных) генов. Иногда симптомы липидоза проявляются у пациенток с одним дефектным геном, когда доминантный ген оказывается инактивированным (причины этого не выяснены).
Патогенез
Патогенетической основой большинства липоидозов является генетически детерминированный ферментативный дефект – энзимопатия. Вследствие этого лизосомы клеток накапливают липиды и промежуточные продукты их метаболизма, что ведет к прогрессивному нарастанию нарушения функций органов. При синдроме Вольмана определяется недостаточность кислой этеразы, становится невозможным полный цикл обмена холестерола, повышается содержание его эфиров в лизосомах селезенки, печени, надпочечников, кишечника и костного мозга. У пациентов с болезнью Гоше снижено или полностью отсутствует производство бета-глюкозидазы; в печени, селезенке, костном мозге скапливаются продукты расщепления сфинголипидов. Болезнь Нимана-Пика развивается на базе дефицита сфингомиелазы, характеризуется повышением количества сфингомиелина во многих клетках, особенно в гепатоцитах, нейронах. При идиотии Тэя-Сакса существует дефект N-ацетилгексозаминидазы, происходит накапливание ганглиозидов в головном мозге.
Классификация
Наследственные нарушения метаболизма сложных липидов представлены группой различных по происхождению заболеваний, которые на патогенетическом уровне связаны с патологией жирового обмена. Для липоидозов характерно скопление сложных липидных соединений внутри лизосом клеток. В зависимости от того, какой липид не расщепляется до конца и откладывается в тканях, выделяют несколько типов болезней:
- Гликолипидозы. При данном типе заболеваний невозможен полный распад гликолипидов – соединений, состоящих из углеводов и жирных кислот. Гликолипидозы представлены цереброзидозами (сфинголипидоз Гоше, болезнь Краббе), сульфатидозами (метахромотическая лейкодистрофия), церамидолигозидозами (болезнь Фабри, церамидлактозидлипоидоз), ганглиозидозами (болезнь Сандхоффа, ранняя детская амавротическая идиотия).
- Липопротеинемии. Обусловлены патологией обмена липидов плазмы крови, основанной на генетическом дефекте ферментов или рецепторов клеток. Липиды плазмы – жирные кислоты, триглицериды, холестерин. Липопротеинемии включают семейную гиперхолестеринемию, комбинированную семейную гиперлипидемию и семейные гиперлипидемии типов I-V.
- Сфингомиелиноз. Синонимичное название – болезнь Ниманна-Пика. При развитии этого заболевания в ретикулоэндотелиальных клетках увеличивается содержание фосфолипидного соединения сфингомиелина.
Симптомы липоидозов
Клиническая картина липидозов определяется особенностями вовлечения в патологический процесс органов и систем. При болезни Гоше I типа поражается печень, селезенка, кости и костный мозг. Симптомы включают увеличение размеров печени, хрупкость костей, анемию, лейкопению, снижение свертываемости крови. II тип заболевания разворачивается с преимущественным повреждением ЦНС и печени. Наблюдаются судорожные приступы, мышечный гипертонус, спастичность, интеллектуальная недостаточность, расстройства акта глотания. У людей с галактозилцерамидным липидозом (болезнью Краббе) снижается функциональность миелиновой оболочки. Развивается гипервозбудимость, рвота, судороги, задержка психомоторного развития с прогрессирующим снижением интеллекта и зрения.
При метахромотической лейкодистрофии патологически изменяется миелиновая оболочка. Основные симптомы – гипотония мышц рук и ног, снижение (отсутствие) сухожильных рефлексов, атаксия, атрофия зрительных нервов, нистагм, спастический тетрапарез, глухота, интеллектуальное и моторное недоразвитие. Клинические признаки болезни Андерсона-Фабри разнообразны, одними из наиболее распространенных являются нейропатическая боль (умеренной и легкой выраженности, в ступнях и ладонях) и кризы Фабри – сильные жгучие боли в конечностях приступообразного характера. Дополнительно возникает гипогидроз или ангидроз, непереносимость физических нагрузок, ангиокератомы, расстройства слуха, сердечно-сосудистой и почечной функций.
При развитии синдрома Сандхоффа определяется общая вялость, гипотонус мышц конечностей, трудности сосания и глотания, прогрессирующая задержка моторного и психического развития, двигательная слабость, шумы в сердце, судороги, слепота, увеличение селезенки. При ранней детской амавротической идиотии больше всего поражается центральная нервная система. К концу первого полугодия ухудшается реакция детей на внешние сигналы (лица близких, игрушки), утрачиваются двигательные навыки, снижается познавательный и игровой интерес. Нарушается зрение вплоть до слепоты. Формируются судорожные припадки.
Первые симптомы сфингомиелиноза – вялость, малоподвижность, апатичность, отказ от еды, рвота. Позже увеличивается живот (гепатомегалия), конечности становятся худыми, кожа приобретает коричневатый оттенок, периоды заторможенности сменяются гипервозбуждением. Дети отстают в психофизическом развитии. Диагностируется умеренная гидроцефалия, гипертермия, спастический парез ног и рук, приступообразная асфиксия.
Проявления гиперлипопротеинемий обнаруживаются в возрасте до 10 лет. Для всех форм болезней свойственно отложение подкожного жира в виде ксантом, а также абдоминальные боли, панкреатит, гепатоспленомегалия (скопление липидов в селезенке, печени). При комбинированной семейной гиперлипидемии и гиперлипидемии II типа возможен ранний атеросклероз сосудов. При гиперлипидемиях IV и V типа – снижение чувствительности к глюкозе, ИБС.
Осложнения
Липоидозы протекают с развитием недостаточности жизненно важных органов. Наиболее частыми осложнениями становятся заболевания сердца и сосудов, ЦНС, почек, легких, печени. Пациенты страдают от атеросклероза сосудов, сердечной и дыхательной недостаточности, ишемической болезни сердца, хронической почечной и печеночной недостаточности, аденом и цирроза печени, инсультов, транзиторных ишемических атак. При типах заболеваний, совместимых с жизнью, зачастую нарушено двигательное и психическое развитие. Многие больные оказываются неспособны к самообслуживанию, обучению и овладению профессией, нуждаются в пожизненном уходе со стороны окружающих.
Диагностика
Липоидозы не являются узкоспециализированными заболеваниями, поэтому их выявлением и лечением занимаются педиатры, гематологи, гастроэнтерологи, ревматологи, неврологи, психиатры и врачи-генетики. На первичном этапе диагностики проводится сбор семейного анамнеза: при наследственном характере ферментопатии у больного могут быть родственники с подтвержденным диагнозом липидоза. При сборе клинических данных специалисты обращают внимание на время начала симптомов: чаще всего болезнь проявляется в период новорожденности или первого года жизни, редко – у детей постарше или взрослых. К специфическим методам обследования пациентов относят:
- Анализ активности дефектного фермента. Исследованию подвергаются различные биоматериалы – плазма крови, лейкоциты, сухие пятна крови, культура фибробластов кожи, биопсийный материал почек, печени. При липоидозах определяется недостаток активности определенного фермента: от легкого снижения до полного отсутствия.
- Количественное исследование липидов. В крови и биопсийном материале органов анализируется содержание патологически накапливаемых липидов и промежуточных продуктов их обмена. У больных липоидозом показатели превышают норму. Параллельно изучаются изменения строения пораженных клеток.
- Секвенирование ДНК. Выявление дефектного гена в хромосоме является наиболее точным, но трудоемким методом диагностики наследственных болезней. Широко применяется в рамках перинатальной и преимплантационной диагностики, в случаях, когда вышеперечисленные анализы не дают однозначного представления о диагнозе.
- Визуализирующие исследования органов. Дополнительно проводится диагностика состояния пораженных органов – сердца, печени, желчного пузыря, селезенки, легких, почек, головного мозга. Используется УЗИ, МРТ, КТ, ЭКГ, ЭЭГ. Процедуры позволяют оценить размеры, выявить структурные изменения и новообразования в органе.
Лечение липоидозов
Терапия данной группы заболеваний – сложная задача для врачей разных специальностей. Методы лечения несовершенны и продолжают разрабатываться, при некоторых видах болезней возможно добиться лишь временного улучшения самочувствия больного, при других достижима стойкая ремиссия. Общая схема медицинской помощи больным состоит из трех компонентов:
- Ферментозаместительная терапия. В организм пациентов вводятся препараты с искусственно выделенным дефицитарным ферментом. Инъекции выполняются пожизненно, позволяют восстановить метаболизм липидов.
- Субстратредуцирующая терапия. Лечение направлено на снижение интенсивности образования патологически накапливаемого соединения (сложного липида, его метаболитов). Используются молекулы с низкой массой, которые стимулируют остаточную активность фермента.
- Симптоматическая терапия. Препараты подбираются индивидуально на основании клинической картины болезни. Распространено применение антиконвульсантов, обезболивающих средств, ингибиторов АПФ, гепатопротекторов. При отдельных видах липоидозов использование симптоматических лекарств является единственным способом лечения.
Прогноз и профилактика
Липоидозы характеризуются гетерогенностью генетического дефекта и выраженной клинической полиморфностью. Большинство из них имеет непрерывное прогрессирующее течение. Правильная диагностика и своевременное лечение позволяют увеличить продолжительность жизни больных, а при легких формах способствуют улучшению адаптации. Профилактика возможна на этапе планирования и в первые месяцы беременности. Супружеским парам с высоким риском передачи болезни ребенку рекомендуется медико-генетическое консультирование, а в первом триместре – исследование амниотической жидкости и материала биопсии хориона на наличие мутаций генов.
| Литература1. Лизосомные болезни накопления липидов у детей/ Захарова И.Н. и др.// Медицинский совет. — 2016 — №1.2. Клиническая генетика/ Давиденкова Е.Ф., Либерман И.С. — 1975.3. Наследственные нарушения обмена липидов: учебно-методическое пособие/ Якимович Н.И. — 2016. | Код МКБ-10E75 |
Вконтакте
Google+
LiveJournal
Одноклассники
Мой мир
mukpomup.ru
липидоз — это… Что такое липидоз?
липидоз — (lipidosis; липиды + оз) 1) (липоидоз устар.) общее название наследственных или приобретенных болезней, характеризующихся нарушением липидного обмена; 2) см. Дистрофия жировая … Большой медицинский словарь
Липидоз (Lipidosis), Липоидоз (Lipoidosis) — нарушение внутриклеточного липидного обмена в организме. Церебральный липидоз (brain lipidoses) (см. Болезнь Гоше, Синдром Гунтера, Синдром Гурлера, Болезнь Тэя Сакса) является врожденным нарушением внутриклеточного липидного метаболизма,… … Медицинские термины
ЛИПИДОЗ (LIPIDOSIS), ЛИПОИДОЗ — (lipoidosis) нарушение внутриклеточного липидного обмена в организме. Церебральный липидоз (brain lipidoses) (см. Болезнь Гоше, Синдром Гунтера, Синдром Гурлера, Болезнь Тэя Сакса) является врожденным нарушением внутриклеточного липидного… … Толковый словарь по медицине
фосфатидный липидоз — Nieman Pick disease болезнь Ниманна Пика, фосфатидный липидоз. HЗЧ из группы липидозов, характеризуется увеличением печени, селезенки, лимфатических узлов, прогрессирующим исхуданием, наличием в кроветворных органах фосфатид позитивных крупных… … Молекулярная биология и генетика. Толковый словарь.
Липидо́зы — (lipidoses; липид [ы] (Липиды) + ōsis) группа заболеваний, характеризующихся нарушением липидного обмена и имеющих преимущественно наследственный характер. Большинство Л. относится к болезням накопления (Болезни накопления), которые обусловлены… … Медицинская энциклопедия
Тилорон — (Tiloronum) Химическое соединение … Википедия
болезнь Ниманна-Пика — фосфатидный липидоз HЗЧ из группы липидозов, характеризуется увеличением печени, селезенки, лимфатических узлов, прогрессирующим исхуданием, наличием в кроветворных органах фосфатид позитивных крупных вакуолизированных клеток, умственной… … Справочник технического переводчика
дистрофия жировая — (d. adiposa; син.: декомпозиция жировая, Д. липидная, липидоз, липофанероз, ожирение дегенеративное, ожирение декомпозиционное, фанероз жировой, фетофанероз) Д., характеризующаяся нарушением обмена липидов и проявляющаяся либо увеличением… … Большой медицинский словарь
липоидоз — (устар.; lipoidosis; липоиды + оз) см. Липидоз … Большой медицинский словарь
Дистрофия — I Дистрофия у детей (dystrophia; греч. dys + trophē питание) хронические расстройства питания у детей. Различают следующие основные виды дистрофии: гипотрофию, гипостатуру, паратрофию и гипертрофию (см. Ожирение). Кроме того, выделяют особый… … Медицинская энциклопедия
dic.academic.ru